����ۺϣ�Interfacial Polymerization, IP����ˇ���Ƃ�����ܷ��xĤ�����ܱ�Ĥ�͏ͺ�Ĥ���ϵ���Ҫ�������V��������ˮ̎�������ӷ��x���ܽ��昋�����I��Ȼ�����ڽ�������̎�l���ķ����c�Uɢ�F�����s�ҟo���������ֶζ���ҕ����ϻ��W�I���L��δ����y�}֮һ��
���ڣ���ۿƼ���W��ڽ��ڈF��ϼ���������WWilliam A. Goddard III���ڈF�����TEPA������ϩ�尷���c HMDI��4,4''''-���h�����������������������ٷ���ģ��ϵ�y���Y�����ӻ��W�͙C���W�������ķ��ӌ���ģ�M�A�y�͌���^�y��C���p�ֶΣ��C����ˮ�����ڽ��淴������Ч�����|���D���܉��Ĵ��C���ͷ����Uɢ·����������������̎���������W�C���c�����{�ؾۺ���Ĥ���^��ò�ṩ������������
2026��3��16�գ����P�о��ɹ��� ��Interfacial Polymerization of TEPA and HMDI: The Role of Water�� ���}�l���ڴ��I��피��ڿ� ACS Catalysis�ϡ����µ���ͬ��һ��������������ʿ����Ӻ����ʿ�����µ�ͨӍ������William A. Goddard III����, �������������ͨӍ��λ�����������WԺ����ۿƼ���W��

���İl�F
���о��Y�����ӻ��WӋ�㡢���ӄ����Wģ�M�c���������ķ��ӳ߶Ƚ�ʾ��ˮ�ڽ���ۺ��е��P�I���á��о��l�F����ˮ���Ӳ���ֻ�ǽ���h����һ���֣������܉�ֱ�Ӆ��c�����^�̡�ˮ���ӿ��ڰ����c����������֮�g�������I�������@�������|���D���܉����Ķ��ӿ����ۺϷ������о��U���˷������A���ڰl�����ЙC��һ�ȵķ���ԭ��ģ�M�c���Y����ͬ������HMDI ��������ʮ�����࣬�� TEPA ������������w�Ʋ��l���������@�Q���˾ۺ���Ҫ���ЙC�������M��ԓ��������Փ�͌��ɷ����ʾ��ˮ���M����ۺϡ�����h���{�ط���λ�õı��|�C�ƣ���Ĥ���Ͻ���ۺ��^�̵Ą����W�{�ء��Y���OӋ�c��ò�����ṩ���µ���Փҕ�ǡ��@һ�l�F�f���������h���е�ˮ���HӰ푷������ʣ�Ҳ�����Mһ��Ӱ�Ĥ�ӵ��γ�λ�á��Y�������Ժ���K��ò��
�D�Ŀ��
���ʾ TEPA �c HMDI ����ۺ��е��|���D�ƙC�ƣ��������Ƚ����˾��д����Եķ���ģ�͡��D1չʾ�� TEPA��HMDI ����ʼ���g�w�Y���������xȡ�� TEPA �IJ������ٰ�λ�c���Լ� HMDI �ăɷN���͘��ͣ����ڱ�������ۺ��п��ܵķ������Ρ��ڴ˻��A�ϣ������Mһ�����^�˟oˮ�l���µ�ֱ���|���D���c��ˮ���Ӆ��c�Ę���|���D�ƃɗl·�����D2�����˱����о�����Ҫ����ģ�ͣ����ڷ���ˮ�Ƿ��܉��ڰ����c����������֮�g�䮔���|���D�Ƙ���������Ҫָ�����ǣ����IJ�δ���]�����������cˮ��ֱ�ӷ���������@һ·���܉��^�ߣ��ڳ�Ҏ�l�����y�l����

Figure 1. Molecular structures of the IP process studied in this work. (a) Molecular structure of TEPA. (b-d) Three conformers of HMDI: (b)trans-trans-HMDI, (c)cis-trans-HMDI, and (d)cis-cis-HMDI. (e) Representative intermediate formed in the initial step of the reaction between TEPA and HMDI, which ultimately leads to a cross-linked polymer network.
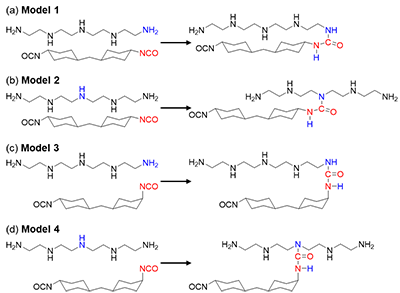
Figure 2. Schematic of the reactant and product for the direct proton transfer reaction between TEPA and HMDI investigated. (a) Primary amine of TEPA with trans-trans-HMDI (Model 1), (b) Secondary amine of TEPA with trans-trans-HMDI (Model 2), (c) Primary amine of TEPA with cis-trans-HMDI (Model 3), and (d) Secondary amine with cis-trans-HMDI (Model 4).
����ϵ�y���^�� TEPA �c HMDI �ڲ�ͬ�h���µ��|���D��·�����Y���������oˮ�l���µ�ֱ���|���D���܉��^�ߣ�������ˮ�h���и��y�l�����@�c����н���ۺϿ����M�еĬF��һ�¡����֮�£�����ˮ���Ӆ��c�r���|���D���܉��@�����ͣ�������������������������Ҳ�����f��ˮ�����mȻ������������s���ڰ����c����������֮�g�䮔��Ч�����|�ӂ��f���������Ķ�ʹ����ۺ����Ҝ��¿��ٰl�������⣬��ͬ HMDI ���ͺ� TEPA ��λ�c֮�g�IJ�������@���f�������Q������Ч�ʵ��P�I�����Ƿ��Ә��ͱ���������ˮ�Ƿ��c���|���D���^�̡�

Figure 3. Calculated reaction pathways between TEPA and HMDI. (a, b) Electron-pushing mechanisms for the direct and single water molecule assisted proton transfer, respectively. (c)-(e) Free energy profiles of direct proton transfer in the gas phase, implicit hexadecane, and implicit water, respectively. (R, TS, and P represent reactant complexes, transition states and products, respectively. (f)-(h) Free energy profiles of single water molecule assisted proton transfer. The energies for separated reactants are available in Table S3 to Table S6. Additionally, the energies for reactions (c)-(h) and their corresponding cartesian coordinates are provided in Table S14 to Table S17, respectively.
�����Mһ�������˸����@ʽˮ���ӵ�Ӱ푡��Y��������ֱ���|���D��ʼ�K���������ˮ���c�r�������@�����װl�������w�������ֲ�ˮ�h��Խ�S����Խ�����ڷ����^�ɑB�����M�|�ӂ��f��ͬ�r���`��У���Y���@ʾ�@һڅ�ݾ����^�߿ɿ��ԡ��D4�Mһ���f����ˮ���H����������|�Ә������c�������܇����^�܄����h��Ҳ�����m��������M���á�
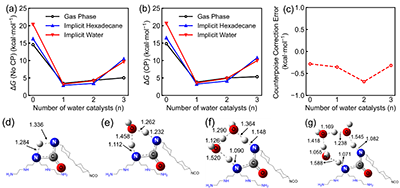
Figure 4. Influence of the number of water catalysts and counterpoise (CP) correction on ?G. (a) ?G for the reaction between TEPA (secondary amine) and HMDI (trans-trans-HMDI) without CP corrections. (b) ?G with CP. (c) Energy differences between results without and with CP correction. (d-g) Transition state structures for the four investigated cases in the gas phase.
ˮ������@�����M�|���D�ƣ������Mһ�������˷����еĚ��I�Y�����Y���������oˮ�l���µĚ��I�����^�����������|�Ӹ�Ч�w�ƣ�����ˮ���Ӆ��c���ڷ�����֮�g������������ӽ����똋�͵Ě��I�Ķ��γɸ������Ă��f·�������ӄ����W�Y��Ҳ�@ʾ��TEPA �İ����������cˮ�γɷ������I���������c��ʮ�����γ��������á��@�f�����ڽ���ۺ��^���У��������M�����M�е��ǽ��渽��ˮ�����������Ě��I�W�j��
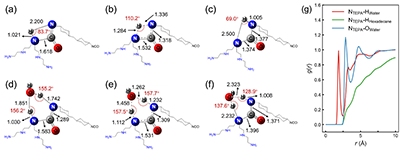
Figure 5. Hydrogen bond structures and solvation analysis for the TEPA-HMDI reaction. (a)-(c) Optimized geometries of the reactant, transition state, and product for direct proton transfer. (d)-(f) Optimized geometries of the reactant, transition state, , and product for single water molecule assisted proton transfer. (g) Radial distribution functions for atom pairs in the water/hexadecane solvated systems.
����ۺϞ�θ��A��l������ʮ������һ�ȣ����ߏķ���������ܳ��l�������� TEPA �� HMDI ��ˮ���c�ЙC���еķ����О顣�����{��� MACE �C���W������ģ�M�Y����TEPA �cˮ������ø������� HMDI �t��ƫ����ʮ�����ࡣ�@��ζ�����ڽ��渽������������ TEPA ��ˮ�����ЙC���ȿ������c HMDI �l�������������� HMDI �M��ˮ�ࡣ��ˣ��D6�ķ��ӌ������˞�ξۺϸ��A��������ʮ��������M��

Figure 6. Accuracy of the fine-tuned MACE FF and calculated interaction energies. Comparison of predicted forces (a) and energies (b) between the Original MACE-MPA-0 model and DFT references. Comparison of predicted forces (c) and energies (d) between the fine-tuned MACE-MPA-0 model and DFT references, the corresponding MAE and RMSE values are provided. (e) Interaction energies between TEPA or HMDI and H2O or hexadecane molecules.
�����Mһ���Y�Ϸ��ӄ����Wģ�M�c�ҵη����揈�����ķ����w���c�����О�ɂ�������C�˾ۺ�λ�á��Y��������TEPA ��������ˮ���з�ɢ��������w�ƣ��� HMDI ��Ҫͣ�����ЙC��һ�ȡ��@һ�ֲ��c�Uɢ�����f��������ۺϲ�������ˮ���Ȳ��l�������Ǹ����ܼ����ڿ�����ʮ����һ�ȵĽ���^���cǰ�����Փ������һ�¡�
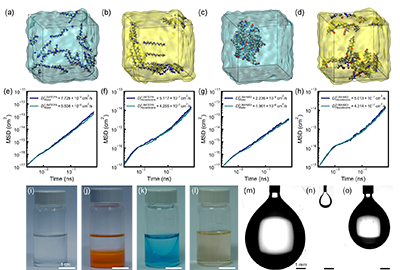
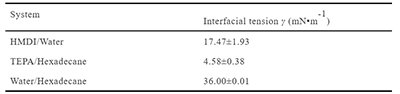
Figure 7. Simulation and experiment-based diffusion and solubility behaviors of TEPA and HMDI in water and hexadecane. (a)-(d) Solute morphologies of TEPA in water, TEPA in hexadecane, HMDI in water, and HMDI in hexadecane studied by MD simulations, respectively. (e)-(h) Mean square displacement curves and corresponding diffusion coefficients of TEPA and HMDI in water and hexadecane obtained from MD simulations. (i)-(l) Experimental observations of solubility behavior. (i) TEPA is completely miscible in water, forming a homogeneous solution. (j) TEPA exhibits phase separation in hexadecane, with hexadecane in the upper layer and TEPA in the lower layer. (k) HMDI forms a hemispherical aggregate in water, surrounded by hydration shells. (l) HMDI is homogeneously dissolved in liquid hexadecane. (m)-(n) Pendant-drop images used for interfacial tension measurements. (m) Water drop in hexadecane, (n) TEPA drop in hexadecane, and (o) HMDI drop in water.
Table 2. Interfacial tensions measured by pendant-drop method at 298.15 K
����ϣ����������@���ɹ��������،�ֱ���^���˽���ۺϵİl��λ���c���ڷ����^�̡��Y����������ˮ�l���½��渽����Ѹ���γ����@�����^���Ҿۺ���Ҫ�l��������һ�ȣ����oˮ�l�������l�����@�ۺϡ��@�f����ˮ�ڽ���ۺ��в��H�����ھS�ַ������棬����ͨ�^���I����ֱ������ TEPA �c HMDI �ķ������ԡ������ڷ�����ʼ�A�Σ��wϵ���� 100 ms �ȿ��ٳ��z���Mһ����������ˮ���ӌ� TEPA �Ļ���ã��Ǽ��ٽ���ۺϵ���Ҫԭ��

Figure 8. Overview of the formation of interfacial polyurea. (a)-(b) Microscopic and fluorescence images of polyurea formation and clear growth after 30 mins at the interface of the experimental group. (c)-(d) Microscopic and fluorescence images of polyurea formation and unclear growth after 30 mins at the interface of the control group. (e)-(h) Early formation process of polyurea upon direct mixing of 99 wt% TEPA and 50 wt% HMDI with increasing time. The scale bars in panels (b) to (h) are identical to that in (a). The arrows in (a)-(d) and (h) indicates the polymer growth direction.
�Y��չ��
���о��Y�����ӻ��WӋ�㡢���ӄ����Wģ�M�͌�������ϵ�y��ʾ�� TEPA �c HMDI ����ۺϵķ��әC�ơ��Y��������ˮ�����܉��@�������|���D���܉����䮔�B�Ӱ����c�������������P�I�����������Ķ����M�Ҝ��µĿ��پۺϡ�ͬ�r��ģ�M�Y���@ʾ��TEPA ������ˮ�����ЙC���ȿ������� HMDI ���A��ͣ������ʮ�������У��@�ķ��ӌ������˾ۺϸ����ܰl���ڿ�������һ�ȵĽ���^���Y��Ҳ�Mһ����C���@һ�c���ۺ���Ҫ���F�������ȣ�����ˮ�o���±��F������ķ������ʺ����@�����z�γɡ����w���ԣ�ԓ������ʾ�� ��ˮ���M�����������ȳ�Ĥ�� ���P�I�C�ƣ����w���ԣ�ԓ������ʾ�˽���ۺ�Ĥ�γ��^������ˮ���M�����������ȳ�Ĥ�����P�I�C�ƣ��������/�������ۺ�Ĥ���������ܱ�Ĥ���ϵķ��������W�{�ء�����Y���OӋ��Ĥ����ò�����ṩ���µ���Փҕ�ǡ�
ԭ��朽ӣ�https://pubs.acs.org/doi/10.1021/acscatal.5c08183
- ��ۿƼ���W��ڽ��ڈF� Adv. Mater.���C���W���o����������ۺ��z�һ��Ķ��������OӋ��ʽ 2026-01-25
- �㽭��WФ�J�о��T�F� AFM���C���W���o��4D��ӡҺ�������w�ͺϽY�����������OӋ 2025-12-13
- �͵���W�ï�n�}�M Angew��������ָ�y - �C���W���c�����{��֮�� 2025-10-31
- ɽ�|�r��/����b�F� AFM������ۺ��^���������Hˮ�Ԍ�Ĥ�γɄ����W�Ķ�Ƕ�Ӱ� 2026-02-11
- �|�A��W������/�ǻ����F� Nat. Commun.�������܄�/��N����ۺ������Ƃ�����ܾ������ЙC�܄��{�VĤ 2025-09-26
- �A�ƴ�/������ Adv. Mater.������������ۺ� - COP Ĥ�ܿ� 2D ���L��ͨ��ƽ�_ 2025-07-16
