�����书�ܡ��ؓ�Y�����B�ӷ�ʽ�Ķ����ԣ�COFs�ѳɞ�Y���OӋ���ܶ��Ƶ��ЙC���ܲ��ϵď���ƽ�_������ʮ�����COFs���S���I�������˻��W�҂��O����dȤ��������w�ă���ͷ��x����������늳ء����ȡ�Ȼ�����������S�����Բ��H�c�����ăȲ��B�ӻ��W�Ͷ�Y�����P��߀�c�����ļ{���ΑB���P�����y���܄���ϳɷ�����Ҫ�^�ߵĜضȡ���ղ������^�L�ķ����r�g���@�����ˌ�COFs����ò���ƺ�Ҏģ�����á��M��Խ��Խ����µĺϳɷ���������������@ʾ�������ڸ�Ч�ϳ�COFs����ă��ݣ�����Ό��F�A���OӋ�Ϳ�����ò��Ȼ��һ������
���˿���COFs���ΑB��Ӳģ�巨����ģ�巨��Ŀǰ��������ΑB�{�ط�������ģ�巨��Ҫ���Ɔ��w�Y���ͷ����l���������˺ϳɸ߽Y����COFs���y�ȡ�Ӳģ�巨��Ҫ�x����m��Ӳģ�壬�����Ӳģ�壬Ȼ���Ƴ�ģ�壬�����^�̲������E���ͣ��Ƴ�ģ����^����߀���ƉĽY���ԡ����H��ܛģ�巨�Ǹ�������Ч���F��ò�{������������ܛģ����Ժ�����ͨ�^ϴ��ȥ����ͨ����ʹ�ñ�����Ԅ�����ܛģ�����Һ�ۺϿ����ڜغ͵ėl���¿��ƾۺ�����ΑB��Ȼ������Һ�ۺϷ��Ƃ��ΑB�ɿص�COFs�Ĉ�����١��@�����ڴ����COFs�ĺϳ���Ҫ���Դ����ʹ������ЙC�܄����@��K�˱�����Ԅ����ΑB�Ŀ��ơ���ˣ��б�Ҫ�_�lһ�N�µ���Һ�ۺϲ��ԣ�����Ч��ʹ������Ԅ�����ܛģ�壬�ڜغ͵ėl���ºϳɾ��п����ΑB��COFs��


��Һ�ۺϷ��Ƃ�Tp-COFs
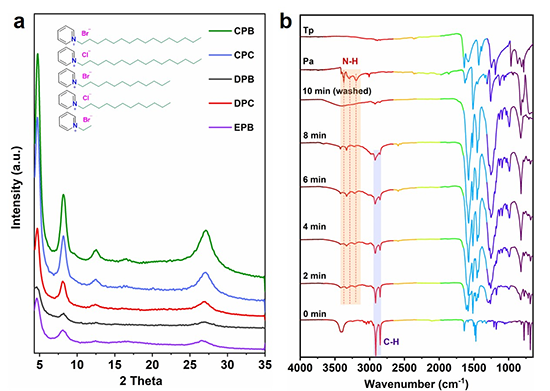
��CPB�����Ч�������Mһ�����������������l����TpPa-COF�ϳɵ�Ӱ푡����ȣ�̽ӑ�˴�����Ȍ��Y���ȵ�Ӱ�������Ȟ�1 mg mL-1�r���Y������ߡ������Mһ���˽������܄������ã�����ͬ�܄��M���ˌ��Ȍ��mȻ��׃�܄�����ʹTpPa-COF�γɸ߽Y���ȣ����γ��˲�ͬ����ò����ͬ�ΑB���γ���Ҫ�c�ЙC�܄��cˮ֮�g�Ľ��揈���Լ�Tp���w���ܽ�����P���ڲ�ͬ���܄���Ҳ���ԫ@�ø߱ȱ��档�����l�FTp���w�ļ�������Ӱ푱ȱ���e����������ٶȞ�0.5 mL min-1��
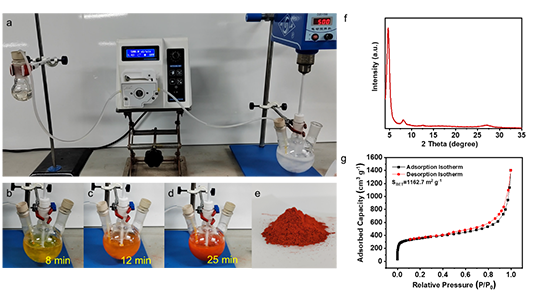
�ڴ˜غͷ��������l���Ļ��A�ϣ��Mһ���C����TpPa-COF�����ڿ������ό��FҎģ���ϳɡ�ͨ�^�Cе����U���wϵ�����Ա��ָ߽Y���Ⱥ�BET����e��1162.7 m2 g-1����Figure 2����ͨ�^�����Ƶĺϳɗl����ʹ�ò�ͬ�İ��������w��Ҳ�C�����@�N�ۺϷ������ձ��ԣ�Figure 3����
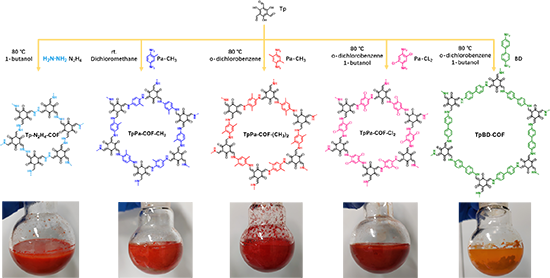
���D�ƴ��ęC���о�
������C�����γɵı�Ҫ�ԣ��M������ˮ�ࡢ�����ȼ�������ˮ-���ȼ��������wϵ�����Ŀ��ƌ��C���������D�ƴ��У�����������Dz���ȱ�ٵġ�ֻ����ˮ-���ȼ�������wϵ�в��܌��F�߽Y���Ⱥ߱���e�ĺϳɡ�ԓ����Ҳ�yԇ��������͵����D�ƴ�������x�ӱ�����Ԅ��������_����������Ч����ֻ��ʹ���L�������D�ƴ������ܫ@�ø߽Y���ȵ�COF���෴���ڛ]�б�����Ԅ�����r�£���ʹ�ڃ����wϵ��Ҳ�]���^�쵽�̶�����ò�ͽY����
���Mһ��̽ӑ���D�ƴ��C�����о��ˆ��w���������COF��ò��Ӱ푡��l�Fֻ���ܽ������Ԅ�������Pa���w���ܫ@�ü{�������ΑB��FT-IR���V�@ʾ��������Ԅ�����Pa���������@׃���������p���{�ƣ��քe��3192��3295��3373 cm-1����3201��3329��3412 cm-1��ͬ�r��CPB��C-N���687 cm-1�{����690 cm-1��Figure 1 b������ˣ��Ɯy������Ԅ������cPa����ã�Scheme 1 step 1��������֮�g���o�����Ì���Pa���z���⾉�����ܣ�Scheme 1 step 2�������ȼ���Ĵ���ʹTp�ܽⲢ�ڷ����г�ַ�����ͬ�r������}��x��߀��ͨ�^ż�O�o�����ü���Tp���w�ϵ��һ���CPB�cˮ�������w�Ƶ����࣬�y��Pa�c���Tp�l��������Scheme 1 step 3�����a��ͣ�����z���ȣ����a��ˮ���ڱ�����Ԅ�����ˮ�������z����������������Ԅ��������ˮ�h��ʹ�����D�ƴ���ˮ��Һ�еļ{���z���{����������ɣ��@�����ǿ��ٺϳ��^�̵���Ҫԭ����10 min��Y�������a��ϴ���CPB����ʧ��Figure 1 b�����f�����ڷ����^���ЃH���������ã�ϴ��ص�o�������@Щ����׃�����������ԓ�ϳɷ����������D�ƴ��C����
Tp-COFs���ΑB�{��

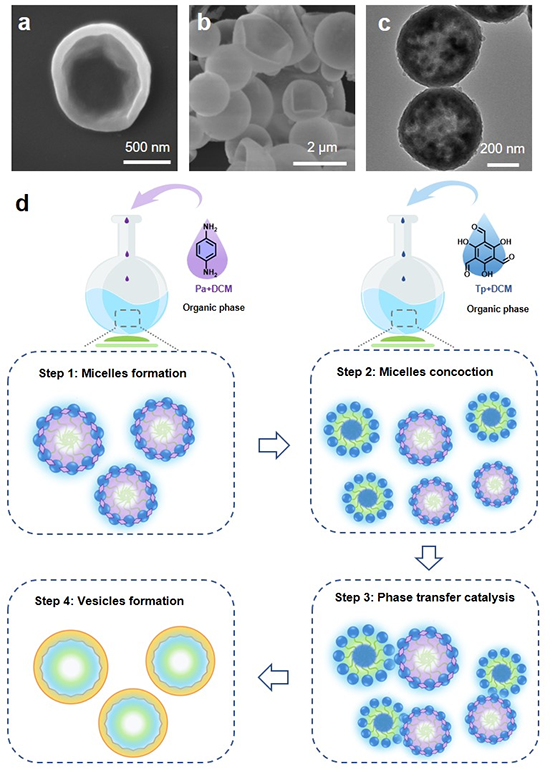
��ͬ�ΑBCOFs�Ĺ������
ԓ�����x����ò��׃��TpPa-COF����ģ��������Mһ��̽ӑ����ò��������ܵ�Ӱ푡���Figure 6 a��ʾ���y���˲�ͬ�ΑBCOFs�Ĺ���������ʣ�HER����e-TpPa-COF���HERֵ��45.8 mmol h-1 g-1�����@���������ΑB���s���w�S�ΑB��5.5 mmol h-1 g-1����8.3��������ΑB��15.4 mmol h-1 g-1����2.97������Ȥ���ǣ�ֱ��ʹ��ԓ�����wϵ���黯Һ���ԫ@�ø��õĮa�����ܣ�HER: 81.6 mmol h-1 g-1������e-TpPa-COF��ĩ��1.78�������r�鄩�������������w���ڱ�����Ԅ�������e-TpPa-COF�������Hˮ�ԣ�����������ˮ��Һ�еķ�ɢ�ԣ�Figure 4 b����

�C��������ԓ�����lչ��һ�N�Gɫ�Ƃ�COF�ķ�����ԓ�������з����ٶȿ졢�ضȵ͡��wϵ���ڷŴ�����c����x�ӱ�����Ԅ�����Һ�ۺ���ͬ�r�����ʹ�����ܛģ������á��@�N�������H�����ڜغ͵ėl���¿��ٴ�Ҏģ�����aCOFs�����ҿ��Կ��Ʋ�ͬ�Π�ͳߴ���ΑB�����⣬COF��Һ����ͨ�^���늈�ֱ���ڌ���r�����Ƃ䱡Ĥ��Ҳ���Ƃ�댧�wCOF��Ĥ�ṩ���µ�;���������������@헹������H���ڜغ͗l���ºϳ�COFs�ṩ��һ�l�µľGɫ;�������Ҟ���N���ڵđ����ṩ��һ�N�µĿ����ΑB�IJ��ԡ�
ԭ��朽ӣ�https://pubs.acs.org/doi/10.1021/jacs.3c06764
- ���A��W�������n�}�M Macromolecules�����w�ܻ��c�����fͬ���Fһ偷��N����Һ�ۺ��Ƃ䲻ͬ��òJanus�w�� 2025-06-11
- �������۽��С��S��/�ɼ{ʿ��WSan H. Thang��Small����RAFT��Һ�ۺ��Ƃ�Ƕ�ι������z�w��Һ�B���Ӿ��w 2025-02-25
- �Ľ�ƽԺʿ�A�ƴ�F�ţȽ�о��T��J. Hazard. Mater.�������ڽ����ЙC��ܵ�ˮ���z�{�R�_����♙z�y��ȥ�� 2024-02-14
- �Ϻ�������W������������V�� Prog. Polym. Sci.����ò�ɿصĶ���ЙC�ۺ��� 2023-05-17
- ɽ�|������W�ǻۭh���F꠳�С����ڡ���W�䲩ʿ�����ܺ���ò�ɿؼ{�VĤ�Ƃ���ȡ�����Mչ 2021-11-27
- �F�ݴ�W�x�m�������n�}�M���܄����l�T������x���Ƃ��^��ò�ɿص�3D�������ײ��� 2018-08-27
- �����Ӣ�����ڡ����ֽ��ڡ�Nano Lett.�������������N�����r�ЙC�Ǽܲ�������������������²��� ���Ŵ������ί�Ч�� 2024-10-21
