�����Қ��w������̼�D����߸���ֵ���ϣ��nj��F̼�к�Ŀ�˵���Ҫ·��֮һ�����գ����ݎ�����W�Ļ��WԺ��������c�㽭��W�߷���ϵ��V���F�����ͨ�^�����OӋ�����f���ܻ����p�����ЙC����������F�˶�����̼�c�h������ĸ�Ч����������lչ�����o���ٴ��wϵ�ṩ����·����
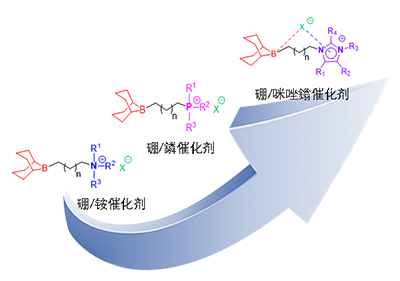
2021�꣬�㽭��W��V���n�}�M�״���������ӃȄӑB·��˹��A��˴��wϵ����DLMCS�����Acc. Chem. Res. 2021, 54, 4434�C4448����ԓ�wϵ�������OӋ�����Ă��P�IҪ�������{�ص�·��˹���������ġ����{�صĿ�����x�ӡ���������x�����Dվ���ļ��@/�l��x�����Լ��B���������c��x�ӵ�����̼朡�ͨ�^���Ӄȅfͬ������ԓ�OӋ��Ч�˷������y�p�M�ִ�����ϡl�������p�����������½����}���Ķ����F�˶�����̼���D���ĸ����c���x�����������@һ����F��lչ��һ�������ԡ�ģ�K�����OӋ�Լ�Ҏģ���Ƃ��������ЙC����wϵ��
���P�о���Copolymerization of CO2 with Epoxides by Imidazolium Engineering in Bifunctional Organoboron Catalysts���}�l����Macromolecules�ϡ����ݎ�����W�Tʿ�о�����ޱ���㽭��W��ʿ�о�������������µĵ�һ���ߣ�������ں���V�����ڞ����µ�ͨӍ���ߡ�
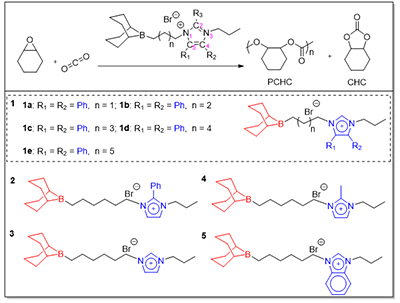
�D1. ��ģ�K���OӋ���ЙC��/�@�f�}���ЙC��/�l�f�}���ЙC��/�����f�}�����
�M�������ш�����ЙC����wϵչ�F�������Ĵ������c���{���ԣ������Y���c����֮�g�Ę�Ч�Pϵ�������nj������Ծ����P�IӰ푵���x�Ӳ��֣���ȱ��ϵ�y���о������磬2022��ԓ�F�ͨ�^ϵ�y������ϵ�c��ϵ�ЙC��������״��C����x���Y���������{���������ܾ��ЛQ����������Macromolecules, 2022, 55, 6443���D1����Ȼ�����oՓ�Ǽ��@�}߀�Ǽ��l�}��x�ӣ������Ч���c���g�Y�����{���ֶ����^�����ޣ�������x�ӵ���������c���wЧ������fͬ�{����Ч������δ�õ�����ϵ�y��������Macromolecules 2024, 57, 8957����
����ǰ���о����A�����ݎ�����W��������c�㽭��W��V���о��n�}�M����������������f��x��������y�@����������x�ӣ�������һϵ�������p�����ЙC������������f��x�Ӿ������ص�����ܗ�Ǽ�����늺��x���������s�h�wϵ����ͨ�^�������벻ͬȡ���������F����������|�c���gλ���ąfͬ�{�����Ķ���ϵ�y��ʾ����x�ӽY���C���������Pϵ�ṩ������ƽ�_��Macromolecules, 2026, 59, 1251�C1261���D1����
�о��F�ͨ�^�{������h��ȡ�����ķN��cλ�����״��U���������f��x��������ܗЧ������x���w���О���{�ؙC���������F�˻���λ�c�g��x���D���^�̵ľ��_�{�أ�ͬ�r���״ΰl�F���������������λЧ���c����h늺�Ч��֮�g��������x�ӵ����������ã�ԓ����ֱ��Ӱ��ۺ�����·���c�����W�О顣���⣬�����������f���е�����ܗЧ����ԓ���չ�F���^�e�ڂ��y���@�}�ͼ��l�}�wϵ�������ۺ��О�����������Ĝض��m�ô�����25�C120 �����c���غ��ɿ��ľۺ����ʣ�TOF > 500 h-1���������������ʾ���p�����ЙC����wϵ����x�ӽM�ֵ��߶ȿ�ģ�K���OӋ���c���{����
�D2. �������о��ĺ������ЙC������Ļ��W�Y��
���о��У��F��OӋ�˃�������������D2����ϵ��һ1a�C1e��ϵ�y�{���������c�����f��x��֮�g�������������L�ȣ�C3�CC7����ϵ����2�C5��������h��C2��C4��C5λ���벻ͬȡ�������������䡢������������������Y��������̼��L�Ȍ������Ծ����@��Ӱ��������������B�ӆ�Ԫ�Ĵ���1d���F�����������L�^�̣�C3�CC5�����^�L��C7�������������½����C���m������g���x�nj��F�������c�����f�fͬ�����P�I�Y��������C2λȡ����Ч��ͻ����C2λ���������Ĵ���2������ߣ��@������δȡ���Ĵ���3������ȡ���Ĵ���4��չ�F�����@���������ԣ������m���Ľo����ԺͿ��gλ�������������������������@�����ƻ��ԣ����������f�Y���Ĵ���5��ʧ��f���^�ȹ�ܗ�����������f��·��˹���Ի���K���P�I���g�w���γɣ��Ķ��ɔ_��ѭ�h��
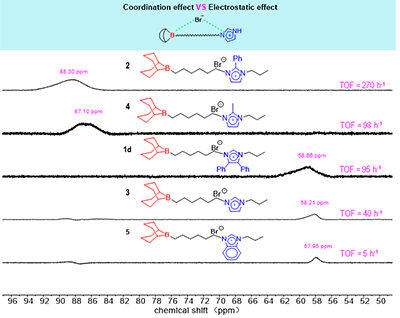
�D3. ����1D��2��3��4��5��CDCl3�е�11B NMR(192 MHz��25��)
11B NMR���Wλ�Ƶ�ϵ�y��׃�����������^�쵽�Ĵ����Բ�ṩ��ֱ�ӵĹ��V�������D3����11B NMR�������������������c�������ĵĻ��Wλ���������P��������ߵĴ���2���� = 88.30 ppm�����F�@���ĵ͈�λ�ƣ���ӳ�������f��x��ͨ�^���Ч���@�������������ĵ�����ܶȡ���������Ҫȡ�Q��ȡ�����T��������ܗЧ�����ɴ����l��������·ֲ������У�N-1λ��������c�����x���������˵�ԭ��������ܶȣ������p���ܴ��wϵ���a�������g��������fЧ����
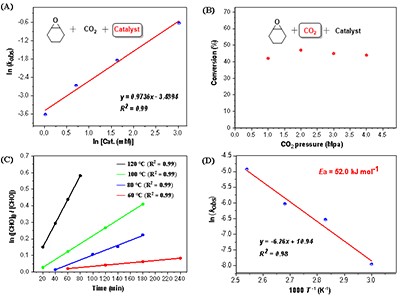
�D4. �����W�yԇ
�����W�о�������������������ȳ�һ����ه����������̼�������㼉��ه���f��������̼������ٿز������h�������ȳ�һ����ه���������^��� (Ea = 52.0 kJ mol-1)���c���ٴ����ஔ���D4����
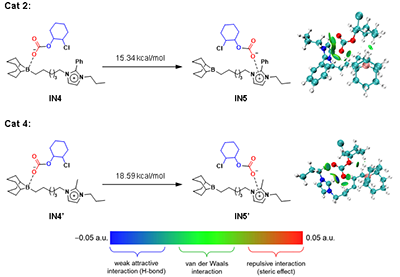
�D5. ����2��4�P�I���g�w��D�Ƶļ���˹������
DFTӋ���Mһ����ʾ������2�б���ȡ������������ܗ��������̼�����x�ӏ��������������f���D���^�̣���G = 15.34 kcal/mol��������ȡ���Ĵ���4�t����ߵ���������G = 18.59 kcal/mol�����Mһ����C�ˌ��Y�����D5����
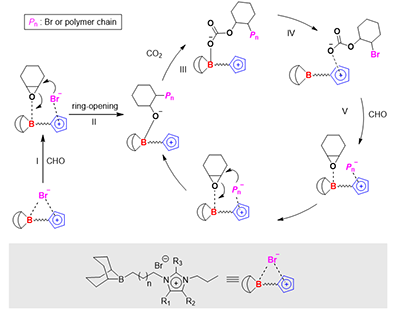
�D6. �������h���h����/������̼���۵ķ����C��
���ڌ���cӋ�㣬�F�������Ӄȅfͬ���C�ƣ��h�������c��������λ��I������Br-�H���_�h��II����������̼�����γ�̼�����x��(III)��̼����D���������f��x��(IV)���ճ��������Ļ��һ���ӭh������(V)��̼����ع��_�h���������L���ۺ��^���������f�䮔����x�����Dվ����ͨ�^�o����Ì���x�������������ĸ������Ķ����F��Ч�fͬ����
���о�ͨ�^ϵ�y�{�������f��x�ӵ�����Y���c���gλ�����U�����p�����ЙC���������x�ӽM���څfͬ��c���w���������P�I���ã���o���ٴ����������OӋ�ṩ���·�ʽ�����_�l�Ĵ������߸��������x�����������᷀���������ڜغ͗l���¸�Ч��������̼�c�h�����w�Ĺ��۷�������ɳ��m��̼�������ϵ��Gɫ�ϳ��ṩ����·����δ����ԓ���wϵ������չ�����N�������h�����w�����F�Y���ɿء����ܿ��{�����ܻ���̼�����ľ��ʺϳɡ�
Փ����Ϣ��Li, W.; Wu, T.; Li, B.; Wu, G.-P. Copolymerization of CO2 with Epoxides by Imidazolium Engineering in Bifunctional Organoboron Catalysts.
Macromolecules 2026, 59 (3), 1251-1261.
https://doi.org/10.1021/acs.macromol.5c03252
