���s�۷eϩ�N��һ��и߷������Ե��H������g�w���䪚�ص���ӽY���ͻ��W���|ʹ���܉�ͨ�^�Mһ���������D�����N�����ЙC������ϳɏ��s�Y���Ĺ��ܷ����ṩ�˶��ӻ��ķ���·�������ЙC�ϳɺ߷��Ӻϳ��I���������Ҫ���о��rֵ�����B������W����n�}�M���ڻ��ڵ��s�۷eϩ�N���g�w��ԭλ�������ۺ��D���_չ��ϵ�y���о���������~�����Ȳ����̼�����������C3ǰ�w�����c�B��ԇ���ĭh�ӳɡ�Ó�������ŵ��^��ԭλ�������s��ϩ�IJ��ԣ����Mһ�����F�����g�w�ژ����Լ��D���еą^�����w�x�����{�أ������Ԍ�����Ƃ��˶�N�Y���S���ľۺ��
���գ�����n�}�M�����һ�Nͨ�^�1,3-��ܗϩȲ�C5ǰ�w��Ȳ��ϩ������������ϩ�����s�۷e��ϩ���g�w�IJ��ԣ���Ч�ϳ������Ѓ���ȫE-�x���Ե�(E, E, E)-��ϩ���������ۺ��
2025��10��30�գ���Nat. Commun �ڿ��ϰl�����}����Vinyl-aza-[3]cumulene intermediates induced polymerization for accessing E-dienyl poly(sulfonylamidines) towards pH-responsive drug delivery�����о�Փ�ģ�Nat. Commun. 2025, 16, 9588�������B������W�������n�}�M���c�˴�헹����������ڴ˹������ͨ�^�{��1,3-��ܗϩȲ�C5ǰ�w����-λ�h�������^���Mһ�����F���a������w�x���Կ��ƣ����x�����Ƃ���(E, Z, E)-��ϩ���������Y����Org. Lett. DOI: 10.1021/acs.orglett.5c04129����

�D1. �о������c����OӋ
���~����Ȳ��ϩ�������h���������x����ȡ�������Ć��l��ͨ�^���Ӿ�����ʹ�õ�ȡ���~�����Ԍ���ϩ��������ϩ���~���g�w�ܺõ��D������ϩ�����s�۷e��ϩ���g�w���Ķ����F��Ȳ��ϩ��������-λ�H�˷������Ը߮a�ʺ��x���Եõ����Ѓ���ȫE-�x���Ե�(E, E, E)-��ϩ�������ߡ������@�NȲ��ϩ���������������B���Ͱ��Ķ�M�ַ�����MCR���ĽY�����n�}�M�_�l��������M�־ۺϷ�����MCP���������Ȳ��ϩ����ֱ���D�����������_���w���͵�E-��ϩ����朵ľۣ���ϩ��-N-�����ߣ�����������Mn�����_26700 g/mol�ġ����⣬�ۣ���ϩ��-N-�������ߣ�ͨ�����F�����õ��ܽ��ԡ��᷀���������W�����ԣ������W�ɽ����ԡ����оۺ��ﶼ����ˮ���ԽM�b�Ɏ�ؓ늺ɵļ{���w����NPs�������ۺ���P1��zeta�λ�Y���@ʾ�ۣ���ϩ��-N-�������ߣ�չ�F�������x�����|���������λ�SpHֵ�����߶��@�����͡������@Щ���ص����|���@Щ�ۺ�����������d��ù�أ�DOX����ˎ�����õ����Ѓ����dˎ����loading 45.1%����DOX-NPs��DOX-NPs���w��ጷŌ�����������@����pH푑����ԡ�
�����xȡȲ��ϩ�������������N-�������͌��ױ������B����TsN?������С����ģ������������^�������l���������_��С����ģ�巴���ėl����CuBr�������DIPEA��A��MeCN���܄��Ҝ��·���4С�r���܉���90%�Įa�ʣ�E:Z > 95:5�����w�x���Եõ�(E, E, E)-��ϩ�������߮a��3a���ҟo�����^���w�����w�����w�a����ͨ�^���S�˴Ź������V�_���a��3a��ϩ�N���ֵ����w���W���ͣ����⣬ͨ�^X�侀�ξ�����Ҳ�C��3b��ϩ�N���͞�ȫ��ʽ�Y�������D2��
ԓ�������ܵęC�����£�һ�r�~���������cĩ��Ȳ�N1b�l������������-��������g�wint-1����DIPEA�����°l��ȥ�|�ӻ����D����Ȳ�~���g�wint-2��int-2�ϵ��������Ѹ���xȥ���F��-λ���h���������γ��P�I����ϩ��������ϩ���~���g�wint-3��ͬ�r���ڻ�׃�����wint-4��int-5�������cTsN?�����γ����g�wint-6��int-7���S��l���_�h���ţ��õ��P�I����ϩ�����s�۷e��ϩ�����g�w�����ڿ��gλ������������ՓӋ����DFT���Y������E-���ͮa����^�ɑB�܉���Z-���͵�4.5 kcal/mol����ˣ����ɱ��H��ԇ�����w�x���Բ��@���õ�(E, E, E)-��ϩ�������߮a�

�D2. С����ģ�巴���c�����C��
��ģ���������A�ϣ�ͨ�^ʹ���p�B���c�p�����w�������Ե���ϩ��-���s�۷e��ϩ������M���ۺϷ����������з�ʽ��ϩ����朵ľۻ����ߡ��ۺ���������l���飺CHCl3�鷴���܄���Ȳ��ϩ�������p�������B�����p���ı�����2.5:1:1�����w��Ȟ�0.5 M��������CuBr��DIPEA���A���Ҝط���16С�r��

�D3. Ȳϩ���������p�B��������p���Ķ�M�־ۺϷ���
���˴_�����Ƃ�ۺ���Ļ��W�Y������P1�M����1H NMR��13C NMR�Լ�MALDI-TOF MS�����������w��1a��4a��5a���cP1�Ě��V�M�б��^���D4A���������^�쵽��P1�У�1a���� 3.31 ppm̎��ĩ��Ȳ�N���4a���� 3.58 ppm̎�Ļ����ʧ��ͬ�r���F��ϩ���|�ӵ��·壨�� 5.96��6.05 ppm�����C���l���ۺϷ������S��5a�D����P1���B�����w�S���|�ӷ��5.21 ppm����5.16 ppm��ͨ�^���Ȇ��w�cP1��̼�V���D4B���������^������P1��̼�V�м��Ҳ���1a��ĩ��Ȳ�N̼�壨�� 79.0��82.0 ppm����Ҳ�Ҳ����c�xȥ���F���P�������壨�� 82.7��157.7 ppm������P1�У���s�� 24.7-65.2 ppm̎�����^�쵽���Ԇ��w4a��5a�����������̼����̖��ͬ�r���� 165.4 ppm̎���F��һ���µ�C=N�I�塣�����C���������N���w1a/4a/5a�ɹ����c��MCP���������⣬��MALDI-TOF�D���^�쵽һϵ���g����1165.6 Da���x�ӷ壬�@�c�ۺ���P1�؏͆�Ԫ����Փ������һ�£��D4C����

�D4. �ۺ���ĽY������
�����Mһ���S���ۺ���N���Ȳ��ϩ�������p�����p��/�����p�B��������������w�M�кY�x���D5��������ģ�;ۺ���P1֮�⣬Ȳ��ϩ������R1λ�ÿ��Լ�����������o��ӷ����Լ�����������81-94%�Įa�ʵõ�������������Mn����19100-21100 g/mol�ľ۶�ϩ����P2-P4������P5���ܽ����^��������w��Ȟ�0.1 M�r����94%�Įa�ʵõ�Mn��14200 g/mol��P5�������p�����w��ʹ�þ����^�����朵Ķ��������r����85-92%�Įa�ʵõ��a��P6��P7����Mn��22800-26700 g/mol��Ȼ������ʹ�ö�������r��P8�Įa�ʺ�Mn���@���͡������p�B�����w����ʹ���������B�ӵ��p�����B���r��P9-P11��Mn�ͮa�ʾ��������͡�ֵ��ע����ǣ���ʹ���p�����������w�r���܉�ͨ�^MCP�����õ�����N-H���F�ľۻ�����P12���a�ʞ�70%����������16400 g/mol����ʹ���p�����p�Ӵ����p���r������ʹMCP��������M�У��õ��ہ�������P13-P14��Ȼ�������Á���������ۺ���������朵�ϩ�N�Y���������^�͵�E/Z�x������

�D5. �ϳɵľۺ����
������C�ۺ�����������ԣ��������Եľۺ��P1��P2��P9��P10���M���˟��ط�����TGA���yԇ�����Мyԇ�ľۺ��ﶼ�����^�ߵķֽ�ضȣ�Td5%��258.8-273.9 ��C����������������õğ᷀���ԣ��D 6A������ʾ�������ᷨ��DSC�������@ʾ��P1-P2��P9-P10�IJ������D׃�ضȣ�Tg����56.2��96.8 ��C֮�g���D 6B�������ڴ��ڄ�����朣�P9��Tg��ߡ����֮�£���������朵�P10�͎�������朵�P1��Tg�������͡�ͨ�^���^P1��P2����������ؿ������ۺ����к�������ķ��h������Tg�@�����ߡ�
�����Mһ���о�ԓ�ۺ���Ļ��W�����ԣ��ڲ�ͬ�Ļ��W�h�������� HCl��K2CO3��H2O2���Ќ�P1�M���˷�������C���D6C����P1�����Ի������l����ͨ�����ַ�����Ȼ�����ۺ�������65 ����DMSO/MeOH��1:1�����A�ԗl�����l�����⣬���������������F��ˮ���������F���D6D����36С�r��GPC�y���z�y���ۺ�����̖����ͨ�^���������x�õ���ϩ�����С���Ӯa�

�D6. �ۺ���ğ᷀���Ժͻ��W������
������ϩ��-N-�������Y�����ۺ��������͵ă����x����������D7A��ʾ����DMSO/H2O = 1:9������pH�l���£����оۺ��ﶼ����ؓ늺ɣ��@��Ҫ������N-�������cOH?֮�g�γ��˚��I���Լ��������������������Q���������оۺ��ﶼ����ˮ��Һ���ԽM�b�ɼ{���w����ͨ�^�ӑB��ɢ�䣨DLS���y����������75.6-156.2 nm�����^�״����������ᣨTFA��̎�����^�쵽P1��zeta�λ�������ߣ��D 7B������14.9 ppm TFA�l���£�P1��zeta �λ������-2.3 mV���D 7B�����@�����ۺ���{���w���cOH-֮�g���ښ��I����á���pH = 4�r��P1��zeta�λڅ����0���ʬF�������x�����ԣ��D 7C�������⣬P1��ؓ�λ�S��pH�����߶��@�����ͣ���pH = 11.9�r��zeta�λ�_��-75.2 mV���D 7C�����@�������ډA���ڵ���r�£��ۣ���ϩ�������ߣ��еĻ����߽Y���D׃�鎧ؓ늺ɵĻ�������ͨ�^�z�y���V��6.4-6.6 ppm̎��ϩ�N����̖���_�C�����@һ�c���D 7D���ң����Mһ�����pHֵ��zeta �λ���ٽ��ͣ������^�쵽�г����a�����@�������ɾۺ���ˮ������ġ�
�S�����Î�ؓ늺ɵ�P1ͨ�^�o����ú���ˮ��������x��ˎ���M�����⡣�x��ù�أ�DOX������ģ����ˮˎ����Ў���늺ɵĻ��F���������F�������cP1�ϵ�ؓ늺�����á�ͨ�^DOX�cP1�������Ƃ���ؓ�dDOX�ļ{���w����DOX-NPs����DOX-NPs�ijߴ�������197.2 nm����D7E ��ʾ��DOX-NPs �@ʾ����ˎ��ؓ�d����DLC��15.5 - 45.1%����ˎ��ؓ�dЧ�ʣ�DLE��90.2-92.7%����P1��ˎ��ؓ�d�����ںܴ�̶������ھۺ����cˎ����ӵ��o�����ã��@�N����ÿ��Է����ۺ���-ˎ��ͺ������ˎ��ۼ����Ķ����F�ۺ������ˎ��ؓ�d����
P1���������ԗl���������l���λ���D���D 7C���������o����Üp�����@���܌���ˎ��{���w�����x�Ԍ��Fˎ��ጷš���D7F��ʾ���yԇDOX-NPs-1�ڲ�ͬpH�l���µ�ˎ��ጷ���r�����м{���w�������2С�r�ȶ����F�����@�����lጷ��F��ֵ��ע����ǣ�pHֵ��DOX��DOX-NPs-1�е�ጷ��������@��Ӱ푡���72С�r����pH 5.0��pH 6.0�l���£����^80%��DOX��DOX-NPs-1��ጷų�����������ͬ�r�g�ȣ���pH 7.4��pH 8.0�l���£��քeֻ��33%��21%��DOXጷų������@����DOX��ጷž���pH�����ԡ��@һ�Y����������ڵ�pH�h���������|�ӻ���DOX�c�ۺ���֮�g���o�����Üp�����Լ�������pH�h����DOX�Hˮ������Ҳ��������ጷš�
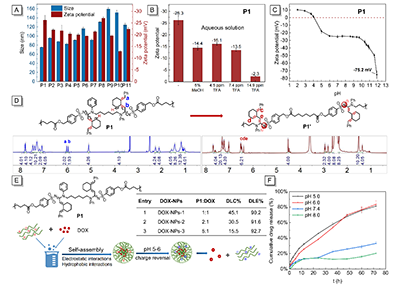
�D7. �ۺ��ԽM�b���dˎ�о�
�C�����������о������һ�N������ϩ�����s�۷e��ϩ���g�w�Ę��������������䑪������M�־ۺ����õ�����(E, E, E)-���w�x���Եľۣ���ϩ��-N-�����ߣ���ͨ�^�~��Ȳ��ϩ�������h�������Լ��c�B�����������h�ӳ�Ó����������ԭλ������ϩ�����s�۷e��ϩ���g�w���P�I���E���õ��ľ۶�ϩ���߲��H�������õğ᷀���Ժͻ��W�����ԣ�߀����ȫˮ����ϩ�������Ԍ��F�ۺ��オ�⡣���⣬ԓ�ۺ���������pH����ؓ�λ����pH = 4�r�l���λ���D������ˮ���ԽM�b�Ƀ����x�Ӽ{���w�����ۺ������ͨ�^�o����ú���ˮ����ؓ�d��ù���Ƃ�����ù�ؼ{������DOX-NPs�������Ѓ�����ˎ��������ܡ��w��ጷŌ�������DOX-NPs���Ѓ�����pH푑��ԣ���pH = 5-6�l���£�72С�r�ȿ�ጷų��^80%��DOX����ˣ�ԓՓ������˸��^���x���Ժ����w�x�����ϳ��������·��������F������Y�����ۺ�����ώ����Y�����ܶ����ԡ�
ԭ��朽ӣ�https://www.nature.com/articles/s41467-025-65320-y
���d��Փ��ԭ����
