方征平,馮煜,金邦梯
(浙江大學高分子復合材料研究所,浙江杭州310027)
Curing kinetics of the binary blends of cyanate ester and epoxy resin
FANG Zheng-ping, FENG Yu, JIN Bang-ti
(Institute of Polymer Composites, Zhejiang University, Hangzhou 310027, China)
Abstract:Curing process of the binary blends consisting of different amount of cyanate ester and epoxy resin was studied by the means of differential scanning calorimetry (DSC) and Fourier transform infrared analysis (FTIR). The apparent activation energy values of CE/EP blends with mass ratio of 10/0, 9/1, 7/3, and 5/5 obtained by ozawa treatment of DSC results equal to 74.3, 72.1, 60.8 and 72.7kJ/mol, respectively. Incorporation of small amount of epoxy resin could accelerate the curing process of cyanate ester. Kinetic parameter obtained by FTIR was in good agreement with that by DSC. Furthermore, it was found that the curing process was a first-order reaction for both cyanate group and epoxy group. During curing process, the transformation of cyanate group was much faster than that of epoxy group, the apparent activation energy values of CE/EP (7/3) calculated by FTIR ways equals to 55.5 kJ/mol.
Key words:cyanate ester;epoxy resin;blend;curing kinetics
摘要:采用DSC和FTIR研究了氰酸酯樹脂/環氧樹脂共混體系的固化行為,考察了環氧樹脂含量對體系的固化動力學參數的影響。純的氰酸酯樹脂及氰酸酯樹脂/環氧樹脂共混物(質量比為9:1,7:3,5:5)的表觀活化能依次為74.3、72.1、60.8、72.7 kJ/mol,說明少量的環氧樹脂可促進氰酸酯樹脂的固化反應,過量則抑制。同時還發現,固化過程中氰酸酯樹脂的轉化速率遠大于環氧樹脂,固化反應對氰酸酯基和環氧基均是一級反應。
關鍵詞:氰酸脂樹脂;環氧樹脂;共混物;固化動力學
中圖分類號:TQ323.9 文獻標識碼:A
文章編號:1001-9731(2004)增刊
1 引言
氰酸酯樹脂(Cyanate Ester Resin, 簡稱CE)性能優異,其介電常數低,使用溫度高,強度高,工藝性能好,是一種具有良好前景的高性能基體樹脂。但是,由于氰酸酯樹脂結構高度對稱且致密,因此較脆,不能很好地滿足使用要求,故有必要對其增韌[1]。為了改善氰酸酯樹脂的韌性,人們提出了許多行之有效的增韌方法。其中用環氧樹脂(EP)與其共固化可降低交聯密度,從而達到增韌的目的。此外,環氧樹脂的價格遠低于氰酸酯樹脂。這樣,采用環氧樹脂增韌氰酸酯樹脂將能大幅度地提高復合材料的性能/價格比,無疑這將對于氰酸酯樹脂的推廣應用有積極的影響。
對于氰酸酯樹脂/環氧樹脂共固化物的性能和固化反應已經有大量的研究[2~4],但是對于其固化動力學的研究限于催化劑如環烷酸鈷/壬基酚的含量對其動力學參數的影響研究,而最重要的影響因素-共混物的組成對其固化動力學參數的影響卻鮮有涉及。因此,本文重點研究氰酸酯樹脂/環氧樹脂二元共混物的組成對其固化動力學影響。
2 實驗
2.1 原料與試劑
雙酚A型氰酸酯樹脂,熔點79°C,純度>98%,中國航空工業第一集團公司濟南特種結構研究所;雙酚A型環氧樹脂,環氧值0.5,江蘇吳江市合力樹脂有限公司;丙酮,分析純,浙江巨化集團公司試劑廠。
2.2 氰酸酯樹脂/環氧樹脂共混物的制備
按配比稱量氰酸酯樹脂與環氧樹脂單體,混合這兩種單體后放在溫度為95℃的水浴中加熱,攪拌10min至均勻熔體備用。實驗中采用的氰酸酯樹脂與環氧樹脂的質量比有以下幾種:10:0、9:1、7:3、5:5。
2.3 示差掃描量熱分析
用PE公司的Pyris-1型熱分析儀測定共混物的示差掃描量熱(DSC)曲線。測定條件如下:升溫速率分別為5℃/min、10℃/min、15℃/min和20℃/min,溫度范圍是25~280℃。
2.4 紅外光譜分析
將氰酸酯樹脂/環氧樹脂(質量比為7:3)共混物用丙酮溶解,溶液涂于KBr鹽片上,置于設定溫度的烘箱中固化,等溫固化的溫度有:160℃、185℃、200℃,固化一定的時間后用德國Bruker公司的Vector-22型傅立葉變換紅外光譜儀測量其紅外光譜(FTIR)。
3 結果與討論
研究熱固性熱固性樹脂固化動力學的常用方法有DSC法、FTIR法、Avrami法等。在本研究中,對氰酸酯/環氧樹脂共混體系的固化動力學用DSC法和FTIR法進行研究。
3.1 示差掃描量熱法研究共混物的非等溫固化動力學
DSC用于研究熱固性樹脂的固化動力學的方法有等溫法與非等溫法兩種。本研究采用非等溫法,其優點是:試樣用量少,測定時間短,測定次數少。測定結果見表1。
從表中可以看出,對于同一體系而言,隨著升溫速率的提高,體系固化的峰值溫度向高溫方向移動。同一升溫速率下的峰值溫度則先是隨著環氧樹脂含量的增加而降低,然后又增加。這可能是由于固化反應中的氰酸酯樹脂與環氧樹脂互為催化劑,固化反應存在最佳配比,此時可以使固化的溫度大為降低,而離該組成比較遠的地方則需要更高的峰值溫度。
對于非等溫DSC法,計算固化反應的表觀活化能主要方法有Kissinger法[5]與Ozawa法[6]。Kissinger法有兩個假定:一是假定反應曲線峰頂溫度處的反應速率最大,二是假定反應為n級反應。這種方法用于DSC研究時,第一個假定基本成立,因為反應曲線峰頂處dH/dt最大,而反應速度dα/dt與dH/dt成正比,也是最大(由下面的研究可知,固化時放熱主要為氰酸酯的貢獻)。但氰酸酯樹脂與環氧樹脂的固化反應很復雜,第二個假定不一定成立。
Ozawa法是一種積分法,不考慮反應機理函數的具體形式。其實驗基礎是:對于同一個固化體系而言,DSC曲線峰頂處的反應程度與升溫速率(r)無關,是一個常數。本實驗中采用Ozawa法對數據進行處理。
根據Ozawa方程: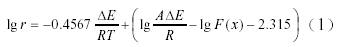
式中F(x)是與轉化率有關的函數,∆E是自由能,R是氣體常數,A是常數。在相同的轉化率下,以lgr 對(1/Tp)(Tp=273.15+ tp)作圖可得一直線(圖1),斜率為k = −0.4567∆E/R,所以表觀活化能∆E為−kR/0.4567,計算結果見表2。
氰酸酯樹脂本身難以熱固化,只有在催化劑的存在下才有較大的反應活性。環氧樹脂的端基為羥基,而羥基上的氫恰恰對氰酸酯樹脂的固化有強烈的催化作用。因此環氧樹脂的加入使固化反應的活化能有較為劇烈的變化。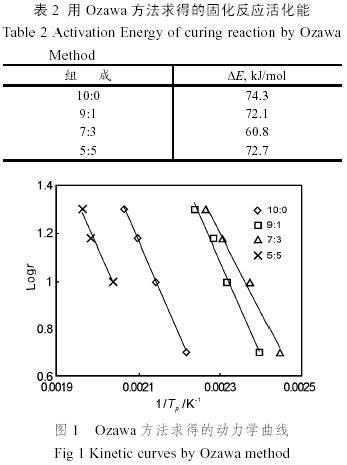
當環氧樹脂的比例由0變為30%時,體系的固化反應活化能大幅度降低,其中環氧樹脂比例為10%的共混物的固化反應活化能與純氰酸酯樹脂相比下降不多可能是由于一些雜質消耗了所加環氧樹脂的活性單元。而環氧樹脂比例繼續增加時,固化反應活化能重新增加,這是因為對于一個需要催化劑的反應來說,催化劑的用量存在一個臨界量,超過該臨界量之后,過量的部分并沒有催化效果。當純氰酸酯樹脂與環氧樹脂作用時,少量的環氧樹脂就足以使氰酸酯樹脂/環氧樹脂共固化體系的氰酸酯樹脂在較低的能量下反應,但在環氧樹脂含量較高時,起催化作用的環氧樹脂的含量并沒有增加,而過量的環氧樹脂固化時又需要較高的能量(這點從下面的FTIR研究中也可以看出來)。因此環氧樹脂含量較高時,氰酸酯樹脂/環氧樹脂共混體系的固化反應的表觀活化能較高。
固化反應的活化能反映了固化反應的難易程度。從表1和2可以看出,氰酸酯樹脂與環氧樹脂的比例對其固化行為有很大的影響。適當地選取兩者的配比,將能大大的降低固化所需要的溫度。
3.2 紅外光譜法研究共混物的固化過程
固化反應的動力學可以根據樹脂的反應性基團的含量隨時間、溫度和固化處理的條件的變化而得到。氰酸酯樹脂/環氧樹脂共混體系在固化過程中的反應非常復雜,對此人們已經作了廣泛而深入的研究,一般認為有以下幾種主要反應: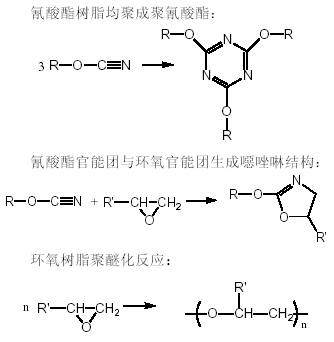
為了使所得到的數據比較準確,采用了在一個溫度點時,同一個鹽片上的樹脂涂層在不同的固化時間點作FTIR,這樣使得同一溫度下的數據有較好的可比性,同時嚴格控制涂層的厚度,使特征峰的高度在整個固化過程中的透過率介于0.2~0.8之間,保證吸光度與濃度之間有較好的線性關系。CE/EP(7/3)共混體系典型的傅立葉變換紅外譜圖見圖2。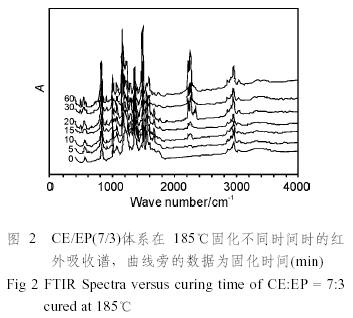
在固化過程中,各個基團的特征峰主要有以下變化:
(1)在1600、1500cm−1為苯環的骨架伸縮振動峰,830 cm−1的峰是苯環的C—H面外彎曲振動峰。在2900 cm−1左右的峰是甲基、亞甲基的對稱和不對稱伸縮振動峰。在固化的過程中,這些峰的高度基本上保持不變,這很大程度上得益于在一個溫度下只使用一塊鹽片,保證了樹脂涂層的厚度不變。
(2)2270 cm−1,2235 cm−1處的兩個強峰是氰酸酯樹脂的氰鍵的伸縮振動峰。在固化的過程中,該峰迅速減弱。而氰酸酯樹脂環化三聚生成的三嗪環(triazine)結構的特征峰(1565 cm−1、1372 cm−1和1120 cm−1)迅速增強。
(3)915 cm−1左右是環氧基的特征吸收峰。該峰的強度在固化過程中逐漸減弱。在1240 cm−1左右有一處較強的吸收峰,它是Ar—O醚鍵伸縮振動特征峰,這說明環氧樹脂的羥基參加了反應。環氧樹脂的羥基與三嗪環反應生成噁唑啉結構,其特征峰在1760 cm−1和1695 cm−1[3]。
3.3 FTIR對CE/EP(7/3)共混體系185℃下固化動力學研究
利用FTIR也可對固化動力學進行定量分析[7],根據Beer-Lambert定理:A=abc
其中A是基團的吸光度,a為基團的吸收率,b為樣品厚度,c為濃度。不同濃度的同一物質在相同波數處具有相同的吸收率,即吸收率沒有濃度依賴性。
對于同一種混合物中的不同波數處的吸光度A1、A2有:
因為對于同一種混合物而言,樣品的厚度是一樣的,而兩者的吸收率之比又是常數。所以: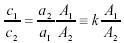 (4)
(4)
在固化過程中,氰酸酯樹脂/環氧樹脂共混體系中的苯環濃度保持不變,可以作為內標基團。如上所述,苯環在1500cm−1和832cm−1處均有特征峰。在本實驗中,選取1500cm−1處的峰為內標,因為該官能團的在固化過程中的峰形與強度隨轉化率變化很小,而且該峰的強度也比較大,作為內標所帶來的計算誤差相對較小。而轉化率α 的定義為: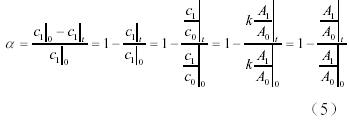
式中的A1和A0分別指待測基團的吸光度和參比基團的吸光度,可以從譜圖上直接測量相應的峰高得到。根據以上各式,我們可以得到共混體系中的氰酸酯基和環氧基的轉化率隨時間的變化,見圖3。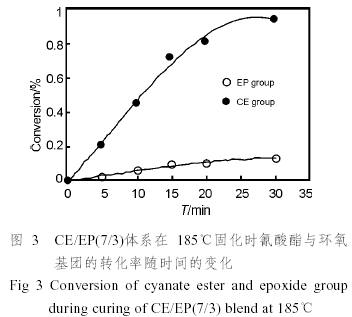
從圖中可以看出,在樹脂固化的早期,氰酸酯樹脂的轉化率很快達到一個較高的值,然而隨著轉化率的提高,轉化率增加趨于緩慢。這是因為體系的粘度變大,分子的運動受阻,交聯反應的速度降低,即反應的決定因素發生了從分子的活化轉化為分子的擴散的轉變,同時未反應的氰酸酯樹脂的量也減少,其反應速率也相應減少。
從圖中還可以得知,在固化的過程中,氰酸酯樹脂的反應速率要遠遠大于環氧樹脂的反應速率,如在185℃反應30min時,氰酸酯樹脂的轉化率已經大于90%,而環氧樹脂的轉化率則仍只有10%左右。
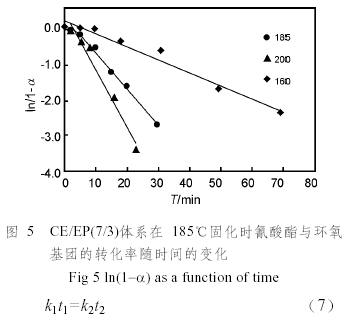
根據不同溫度下的轉化率(圖4),計算各溫度時的ln(1−α),并對時間t作圖,可以得到一系列直線(圖5),因此固化過程中氰酸酯基和環氧基的反應都屬于一級反應。
因為兩者發生的都是一級反應,所以滿足Arrhenius方程: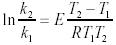 (6)在相同的初始濃度和相同的轉化率時,有下式成立:
(6)在相同的初始濃度和相同的轉化率時,有下式成立: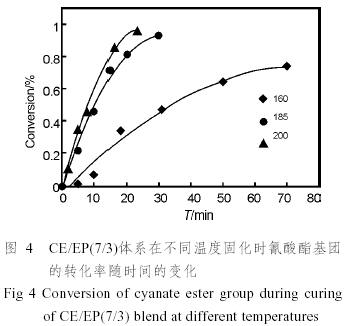

從ln(1−α) ~ t圖中得到固定轉化率時的t和T,取了不同轉化率時的lnt對1/T作圖,得到一系列直線(見圖6),從直線的斜率就可以求得活化能E為55.5kJ/mol。與DSC法得到的活化能60.8 kJ/mol比較接近。
前人的工作[8]發現,對于純的氰酸酯樹脂熱固化,固化反應是一級反應,但是,在催化劑(如環烷酸鈷/壬基酚)存在下,固化反應是二級反應。這與在環氧樹脂催化下的反應是一級反應截然不同。
4 結論
(1)DSC研究表明氰酸酯樹脂/環氧樹脂共混體系的固化過程比較復雜,其動力學參數受氰酸酯樹脂的含量影響較大,氰酸酯樹脂與環氧樹脂的重量比為10:0、9:1、7:3、5:5的體系的表觀活化能依次為74.3、72.1、60.8、72.7 kJ/mol,說明少量的環氧樹脂可促進氰酸酯樹脂的固化反應。
(2)FTIR研究表明在氰酸酯樹脂/環氧樹脂共混體系(7/3)的固化過程中,氰酸酯樹脂的轉化速率遠大于環氧樹脂的轉化速率,固化反應對氰酸酯基和環氧基均是一級反應,其表觀活化能是55.5kJ/mol,與DSC的研究結果比較接近。
參考文獻:
[1] Fang T, Shimp D. A [J]. Prog Polym Sci, 1995, 20: 61-118.
[2] 陳平, 程子霞, 金鎮鎬, 等. [J]. 高分子學報, 2001, (2): 238- 240.
[3] 包建文, 唐邦銘, 陳祥寶. [J]. 高分子學報, 1999, (2): 151- 155.
[4] Kim D S, Shin J H [J]. Polym. Eng. Sci, 2000, 40(6): 1429-1434.
[5] Kissinger E [J]. Anal. Chem, 1957, 29: 1702-1706.
[6] Ozawa, T J, [J]. Thermal Anal, 1970, 2: 301-310.
[7] 沈德言. 紅外光譜在高分子研究中的應用[M]. 北京: 科學出版社, 1982.
[8] Osei-Owusu A, Martin G. C. Gotro J. T. [J]. Polym Eng Sci, 1992, 32(8): 535-541.
作者簡介:方征平(1963-),男,浙江江山人,教授博導,長期從事多組份多相高分子材料的結構與性能研究。(E-mail: zpfang@zju.edu.cn),Tel: 0571-87951295
論文來源:中國功能材料及其應用學術會議,2004年,9月12-16日
