告別OH中毒!武漢理工大學(xué)木士春、李蓓ACS Catal報(bào)道“釜底抽薪”新策略,讓羥基“反水”參與反應(yīng),雙向優(yōu)化HER動(dòng)力學(xué)
原創(chuàng) 研理科研線-文獻(xiàn) 研理科研線-文獻(xiàn) 2025年08月21日 07:30 遼寧
2025年8月13日,武漢理工大學(xué)李蓓、木士春團(tuán)隊(duì)在ACS Catalysis期刊發(fā)表題為“Hydroxyl?Interfacial Water Proton Exchange Strategy to Hasten Water Reduction on Ruthenium Sites”的研究論文,蔣林濤、陳興寶為論文第一作者,李蓓、木士春為論文共同通訊作者。
邏輯鏈條
廣泛背景(堿性HER動(dòng)力學(xué)遲滯)→ 核心問題(傳統(tǒng)觀點(diǎn)認(rèn)為OH中間體吸附會(huì)占據(jù)活性位點(diǎn),“毒化”催化劑)→ 現(xiàn)有方法及其局限(大多致力于促進(jìn)OH脫附,但無法從根本上解決H/OH的競(jìng)爭(zhēng)吸附問題)→ 提出本文核心概念(變“堵”為“疏”,讓吸附的OH物種直接參與反應(yīng)) → 本文策略(通過簡(jiǎn)單的電化學(xué)活化,在Ru表面原位引入活性羥基OHad)→ 闡述假設(shè)的機(jī)制鏈條(Ru-OHad作為質(zhì)子受體 → 啟動(dòng)“羥基-界面水質(zhì)子交換” → 加速水的吸附/解離 → 額外的OH位點(diǎn)促進(jìn)H脫附 → 雙向優(yōu)化HER動(dòng)力學(xué)) → 預(yù)告研究成果(性能大幅提升,并為堿性HER提供了新的反應(yīng)路徑和機(jī)理見解)。
全文點(diǎn)評(píng)
讓我們來看看如何對(duì)這篇文獻(xiàn)深刻的拆解,清晰的呈現(xiàn)到你面前,制作不易,方便的話大家可以點(diǎn)贊轉(zhuǎn)發(fā)喲(公眾號(hào)加上星標(biāo),不迷路喲),尤其建議剛?cè)腴T的研究生同學(xué)可以對(duì)著文獻(xiàn)和我們的解析進(jìn)行對(duì)比閱讀,有利于打開大家的思路,理清邏輯思路。
話不多說,今天這篇文獻(xiàn),它的主角是堿性HER中一個(gè)“臭名昭著”的物種——羥基(OH)。在傳統(tǒng)認(rèn)知里,OH就是一個(gè)“麻煩制造者”,它會(huì)和H搶占催化劑的活性位點(diǎn),導(dǎo)致反應(yīng)動(dòng)力學(xué)急劇下降,這是堿性HER性能遠(yuǎn)不如酸性的核心癥結(jié)之一。因此,過去大量的研究都像是在和OH“打仗”:要么想辦法削弱它和催化劑的結(jié)合,讓它趕快“滾蛋”;要么引入一些親氧位點(diǎn)來加速水的解離,但又容易陷入OH脫附困難的新困境。按理說,這似乎是一個(gè)“死循環(huán)”。那么,作者是如何跳出這個(gè)怪圈,還能在頂刊上講出新故事的呢?這就引出了一個(gè)非常高明的科研思路:打不過,就讓它加入!這篇文章的精髓,就在于它沒有繼續(xù)走“如何趕走OH”的老路,而是反其道而行之,提出一個(gè)大膽的想法:如果OH不是“毒物”,而是反應(yīng)的“催化劑”呢? 作者通過一個(gè)極其簡(jiǎn)單的電化學(xué)活化方法,在Ru表面巧妙地構(gòu)建了一種特殊的活性羥基(Ru-OHad)。這個(gè)OHad不再是一個(gè)被動(dòng)的“占位者”,而是一個(gè)主動(dòng)的“參與者”。
它最絕妙的機(jī)制在于啟動(dòng)了所謂的“羥基-界面水質(zhì)子交換”。你可以把它想象成一個(gè)高效的“中轉(zhuǎn)站”:這個(gè)OHad先從界面水中“搶”來一個(gè)質(zhì)子,幫助水分子順利吸附到催化劑表面并快速解離;同時(shí),多出來的這個(gè)OH位點(diǎn)又反過來幫助生成的H更容易地脫附形成氫氣。這一來一回,不僅完美規(guī)避了OH占據(jù)位點(diǎn)的“中毒”問題,還一舉兩得,從“吸附”和“脫附”兩個(gè)方向上雙重加速了反應(yīng)動(dòng)力學(xué)。
所以說,這篇工作不是簡(jiǎn)單地合成了一個(gè)新材料,而是從根本上挑戰(zhàn)了對(duì)堿性HER機(jī)理的傳統(tǒng)認(rèn)知。它告訴我們,面對(duì)科研中的“頑疾”,有時(shí)候換個(gè)角度,把“敵人”變成“盟友”,反而能柳暗花明。這恰恰體現(xiàn)了科研中從“表象優(yōu)化”到“機(jī)理創(chuàng)新”的深刻轉(zhuǎn)變,為高效堿性HER催化劑的設(shè)計(jì)提供了一條全新的、極具啟發(fā)性的路徑。
理論計(jì)算分析
我們來看這篇文章的DFT理論計(jì)算部分(Figure 5),作者團(tuán)隊(duì)的操作堪稱教科書級(jí)的典范,它沒有停留在簡(jiǎn)單“擬合實(shí)驗(yàn)”的層面,而是從根源上對(duì)催化機(jī)理進(jìn)行了深刻的剖析和預(yù)測(cè)。
首先,作者上來就用一張清晰的示意圖(圖5a)點(diǎn)明了核心科學(xué)問題:活化前后的電子構(gòu)型和反應(yīng)路徑有何不同?這為后續(xù)的計(jì)算指明了方向。整個(gè)計(jì)算邏輯嚴(yán)謹(jǐn),層層遞進(jìn),完美地回答了“為何電化學(xué)活化能極大提升HER性能”這一核心問題:
1. 從“電子結(jié)構(gòu)”探究活化本質(zhì):
功函數(shù)(WF,圖5b)計(jì)算顯示,活化后的R-Ru-Ni(OH)2功函數(shù)更低(3.26 eV vs 3.55 eV),意味著電子更容易脫離表面參與反應(yīng),這與實(shí)驗(yàn)UPS結(jié)果(圖2h)高度吻合。
態(tài)密度(PDOS,圖5c)的計(jì)算更是點(diǎn)睛之筆。它揭示了活化后Ru原子的d帶中心下移,遠(yuǎn)離了費(fèi)米能級(jí)。根據(jù)d帶中心理論,這會(huì)削弱對(duì)中間體(尤其是\H)的吸附,有利于其脫附。這精準(zhǔn)地解釋了實(shí)驗(yàn)中H2-TPD(圖2g)觀察到的更低的脫附溫度。
差分電荷密度(DCD,圖5d,e)和電子定域函數(shù)(ELF,圖5f,g)則將電子轉(zhuǎn)移過程可視化。DCD清晰地展示了電子從Ru核流向Ni(OH)2殼層和表面羥基(Ru-OHad),形成了富電子的羥基位點(diǎn)。而ELF則表明,這個(gè)富電子的Ru-OHad位點(diǎn)具有更強(qiáng)的電子定域性,使其成為一個(gè)優(yōu)異的“質(zhì)子抓手”,為后續(xù)的質(zhì)子交換奠定了基礎(chǔ)。
2. 從“反應(yīng)路徑”驗(yàn)證機(jī)理創(chuàng)新:
吉布斯自由能(ΔG,圖5h)的計(jì)算是整個(gè)理論部分最精彩的一環(huán)。作者對(duì)比了不同反應(yīng)路徑,發(fā)現(xiàn)當(dāng)Ru-OHad作為活性位點(diǎn)時(shí),其與界面水發(fā)生的“質(zhì)子交換”過程能壘極低(僅0.193 eV),遠(yuǎn)優(yōu)于傳統(tǒng)的羥基脫附/水吸附過程。這為作者提出的核心機(jī)理——“羥基-界面水質(zhì)子交換策略”提供了最堅(jiān)實(shí)的理論支撐。
能壘計(jì)算(圖5i)進(jìn)一步量化了這種優(yōu)勢(shì)。計(jì)算表明,Ru-OHad的存在不僅極大地降低了水解離的能壘(僅0.035 eV),更重要的是,它還顯著降低了后續(xù)H脫附形成H2的能壘(0.395 eV)。
總而言之,這篇論文的DFT計(jì)算部分不是對(duì)實(shí)驗(yàn)結(jié)果的簡(jiǎn)單附會(huì),而是一場(chǎng)深刻的理論演繹。它從“電化學(xué)活化引發(fā)電子轉(zhuǎn)移”這一物理現(xiàn)象出發(fā),通過“d帶中心調(diào)控和Ru-OHad位點(diǎn)形成”作為橋梁,最終完美地論證了“質(zhì)子交換策略”如何從“促進(jìn)水吸附/解離”和“加速H?脫附”兩個(gè)方向(Bidirectionally)協(xié)同優(yōu)化了HER的反應(yīng)動(dòng)力學(xué)。整個(gè)論證過程環(huán)環(huán)相扣,邏輯清晰,充分展現(xiàn)了理論計(jì)算在揭示復(fù)雜催化機(jī)理中的強(qiáng)大力量。
摘要解析
摘要是文章的“精華濃縮版”,咱們按照“背景-問題-方案-亮點(diǎn)-意義”的框架來解析:
研究背景: 在堿性析氫反應(yīng)(HER)中,界面水分子(H2O)和反應(yīng)中間體(H、OH)的行為至關(guān)重要。存在的挑戰(zhàn)/問題:傳統(tǒng)的觀點(diǎn)認(rèn)為,OH中間體在催化活性位點(diǎn)上的強(qiáng)吸附會(huì)“毒化”或“堵塞”反應(yīng)位點(diǎn),從而拖慢了整個(gè)HER的反應(yīng)動(dòng)力學(xué)。本研究的方案:作者反其道而行之,提出了一種“羥基-界面水質(zhì)子交換策略”。他們通過簡(jiǎn)單的電化學(xué)活化方法,在釕(Ru)催化劑表面原位引入了被激活的表面吸附羥基(Ru-OHad),意圖讓這個(gè)曾經(jīng)的“麻煩制造者”(OH)變身為反應(yīng)的“催化劑”。
核心亮點(diǎn)與成果:
1. 機(jī)理證實(shí): 通過原位光譜/質(zhì)譜聯(lián)用技術(shù),證明了這些豐富的Ru-OHad物種在HER過程中,確實(shí)建設(shè)性地參與了界面水的質(zhì)子轉(zhuǎn)移過程,從而促進(jìn)了水分子的吸附、裂解以及后續(xù)H?的脫附。
2. 理論支撐: 理論計(jì)算進(jìn)一步揭示,Ru-OHad與H2O的質(zhì)子交換在能量上更有利,同時(shí)Ru-OHad的存在也降低了H的脫附能,實(shí)現(xiàn)了對(duì)HER吸附/脫附動(dòng)力學(xué)的“雙向優(yōu)化”。
3. 性能優(yōu)異: 基于此策略,活化后的催化劑(R-Ru-Ni(OH)2)在堿性介質(zhì)中僅需26 mV的超低過電位即可驅(qū)動(dòng)10 mA cm-2的電流密度,并表現(xiàn)出卓越的穩(wěn)定性。
長(zhǎng)遠(yuǎn)意義:
這種電化學(xué)活化策略對(duì)其他催化劑也具有普適性,為設(shè)計(jì)高效堿性HER催化劑提供了一條全新的、打破常規(guī)的優(yōu)化路徑。
研究亮點(diǎn)與數(shù)據(jù)支撐(證據(jù)鏈)
本研究的核心創(chuàng)新在于,顛覆了傳統(tǒng)認(rèn)知中OH對(duì)HER的抑制作用,巧妙地將其轉(zhuǎn)化為促進(jìn)反應(yīng)的活性物種,并揭示了其通過“質(zhì)子交換”來加速反應(yīng)的全新機(jī)理。 整個(gè)證據(jù)鏈條清晰且有力:
1. 創(chuàng)新點(diǎn):構(gòu)建了活性Ru-OHad位點(diǎn)。
證據(jù)鏈支撐:
XPS光譜(圖2a-c): 清晰地顯示活化后Ru的結(jié)合能正移,Ni的結(jié)合能負(fù)移,證明電子從Ru流向了Ni(OH)2殼層,從而“活化”了Ru表面,使其易于吸附OH。
EPR光譜(圖2e): 活化后的樣品出現(xiàn)了更強(qiáng)的羥基自由基信號(hào),這是存在化學(xué)吸附羥基(OHad)的直接證據(jù)。
物理化學(xué)表征(圖2i, j): 活化后催化劑表面的水接觸角變?yōu)閪0°,展現(xiàn)出超親水性,這與表面富集的親水羥基密切相關(guān)。
2. 核心論證:證明了Ru-OHad通過“質(zhì)子交換”參與并加速HER。
證據(jù)鏈支撐:
原位光譜(ATR-SEIRAS, 圖4c,d): 在反應(yīng)條件下,實(shí)時(shí)捕捉到了歸屬于Ru-OH伸縮振動(dòng)(1013 cm-1)和OH中間體(871 cm-1)的信號(hào)峰,且信號(hào)隨電壓增強(qiáng),證明了Ru-OHad物種真實(shí)地參與了反應(yīng)過程。
同位素標(biāo)記質(zhì)譜(DEMS, 圖4h, i): 這是本研究的“殺手锏證據(jù)”。研究人員先用含D的電解液(KOD/D2O)對(duì)催化劑進(jìn)行活化,使其表面形成Ru-ODad。隨后,將該催化劑放入普通的含H2O的電解液中進(jìn)行HER反應(yīng),質(zhì)譜儀檢測(cè)到了大量的HD氣體產(chǎn)物。這一結(jié)果無可辯駁地證明了:催化劑表面吸附的D(來自Ru-ODad)參與了析氫反應(yīng),唯一的途徑就是通過“Ru-ODad + H2O → Ru-OHad + HDO”這樣的質(zhì)子交換過程。
動(dòng)力學(xué)同位素效應(yīng)(KIE, 圖4g): 活化后催化劑的KIE值(2.413)遠(yuǎn)低于活化前(7.201),表明O-H鍵的斷裂變得更加容易,即水解離過程被顯著加速。
3. 最終效果:實(shí)現(xiàn)了“雙向動(dòng)力學(xué)優(yōu)化”和卓越的催化性能。
證據(jù)鏈支撐:
電化學(xué)性能(圖3): 最終的宏觀表現(xiàn)——26 mV @ 10 mA cm-2的超低過電位和超過100小時(shí)的穩(wěn)定性,證明了該策略的有效性。
DFT理論計(jì)算(圖5h, i): 從理論上完美閉環(huán)了整個(gè)故事,計(jì)算出“質(zhì)子交換”路徑不僅促進(jìn)了水解離(第一步),還降低了H脫附(最后一步)的能壘,為“雙向優(yōu)化”提供了堅(jiān)實(shí)的理論基礎(chǔ)。
前言解析
-
前言部分,作者為我們清晰地勾勒出其研究的邏輯起點(diǎn)和創(chuàng)新思路:
開篇即點(diǎn)明電催化水分解制氫的核心地位,并迅速聚焦于其半反應(yīng)——堿性析氫反應(yīng)(HER)。接著,作者直指當(dāng)前研究的“痛點(diǎn)”:堿性HER動(dòng)力學(xué)遠(yuǎn)慢于酸性條件,其根源在于復(fù)雜的中間體轉(zhuǎn)化過程,尤其是公認(rèn)的OH中間體對(duì)催化活性位點(diǎn)的“占據(jù)效應(yīng)”,它阻礙了關(guān)鍵H中間體的生成和脫附,導(dǎo)致動(dòng)力學(xué)遲滯。作者回顧了現(xiàn)有策略,指出大部分研究都致力于削弱OH的吸附以釋放活性位,但這并未跳出H與OH“競(jìng)爭(zhēng)吸附”的窠臼,從根本上限制了催化性能的提升。
話鋒一轉(zhuǎn),作者亮出了顛覆性的新思路!與其想方設(shè)法“驅(qū)逐”O(jiān)H,不如反其道而行之,利用活化的表面吸附羥基(OHad)來“助推”反應(yīng)。他們提出一個(gè)全新的“羥基-界面水質(zhì)子交換”策略。該策略的核心假設(shè)是:1)活化的OHad作為優(yōu)異的質(zhì)子受體,能有效與界面水分子作用,促進(jìn)水的吸附;2)OHad能直接參與反應(yīng)(OHad + H2O + e- → H2O + OH-),巧妙地繞過了傳統(tǒng)的H2O解離生成H和OH的步驟,從而避免了OH脫附的能壘。這樣一來,OHad的參與從“水的吸附”和“H的脫附”兩個(gè)方向上雙向優(yōu)化了反應(yīng)動(dòng)力學(xué),打破了H與OH的動(dòng)力學(xué)競(jìng)爭(zhēng)壁壘。
最后,作者明確了本研究的工作:通過簡(jiǎn)便的電化學(xué)活化方法,在Ru-Ni(OH)?核殼催化劑的Ru表面原位引入了這種活化的OHad。并計(jì)劃利用原位光譜、同位素標(biāo)記質(zhì)譜(DEMS)和密度泛函理論(DFT)計(jì)算,系統(tǒng)地驗(yàn)證這一創(chuàng)新機(jī)制的有效性。
圖文對(duì)照解析
圖1 材料的合成與結(jié)構(gòu)表征: 成功制備了核殼結(jié)構(gòu)的催化劑,且活化前后形貌保持穩(wěn)定。
-

(a) 展示了催化劑的制備流程示意圖:通過一步水熱法合成Ru-Ni(OH)2核殼納米球,再經(jīng)過電化學(xué)循環(huán)伏安(CV)活化得到R-Ru-Ni(OH)2。
-
(b, c) 掃描電鏡(SEM)圖像顯示,活化前后材料均保持了均一的納米球形貌,表面粗糙。
(d, e) 高分辨透射電鏡(HRTEM)圖像揭示了Ru核-Ni(OH)2殼的結(jié)構(gòu),并表明活化后殼層厚度略有減小。晶格條紋證實(shí)了Ru和Ni(OH)2的存在。
(f, g) 元素分布(EDS)圖譜直觀地證明了Ru元素在核、Ni和O元素在殼的分布特征。
小結(jié): 這一系列表征證明,電化學(xué)活化并未顯著改變材料的宏觀形貌和核殼結(jié)構(gòu),性能的提升來源于更深層次的化學(xué)狀態(tài)變化,而非結(jié)構(gòu)重塑。
圖2 催化劑的化學(xué)與物理性質(zhì): 活化過程成功引入了表面羥基并優(yōu)化了電子結(jié)構(gòu)。
-

(a-c) X射線光電子能譜(XPS)顯示,活化后Ru 3p軌道結(jié)合能正移,Ni 2p軌道負(fù)移,證明電子從Ru核轉(zhuǎn)移到了Ni(OH)2殼層。同時(shí)O 1s譜中代表表面羥基的峰面積增加。
-
(d) 熱重分析(TG)曲線表明,活化后的催化劑在約300°C有更大的質(zhì)量損失,歸因于更多的表面吸附水和羥基物種的脫去。
(e) 電子順磁共振(EPR)譜檢測(cè)到活化后催化劑具有更強(qiáng)的羥基自由基信號(hào),證實(shí)了化學(xué)吸附羥基(OHad)的形成。
(f) DFT計(jì)算表明,在Ru表面吸附3個(gè)OH基團(tuán)(Ru-Ni(OH)2-(OHad)3)的結(jié)構(gòu)在能量上最穩(wěn)定。
(g) 程序升溫氫氣脫附(H2-TPD)曲線顯示,活化后催化劑的H?脫附峰溫更低,表明更有利于H?的脫附。
(h) 紫外光電子能譜(UPS)揭示活化后功函數(shù)降低,d帶中心下移,有利于中間體的脫附。
(i, j) 水接觸角測(cè)試表明,活化后催化劑表面變?yōu)槌H水(接觸角~0°),證明了表面富含親水性羥基。
圖3 堿性析氫(HER)性能評(píng)估: 活化后的催化劑展現(xiàn)出頂尖的HER活性和穩(wěn)定性。
-
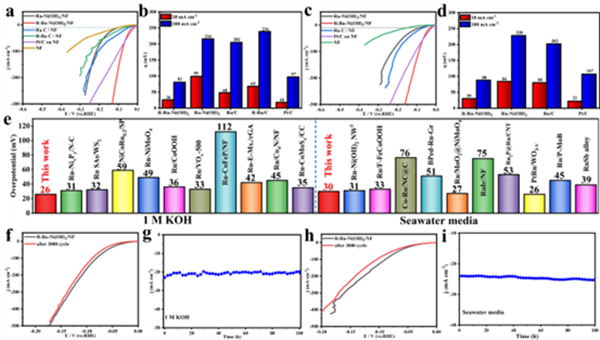
(a, b) 在1 M KOH溶液中,R-Ru-Ni(OH)2/NF僅需26 mV的過電位即可達(dá)到10 mA cm-2的電流密度,性能遠(yuǎn)超活化前樣品和商業(yè)Ru/C,與商業(yè)Pt/C相當(dāng)。
-
(c, d) 在堿性海水中,R-Ru-Ni(OH)2同樣表現(xiàn)出優(yōu)異的性能(30 mV @ 10 mA cm-2)。
(e) 與近期報(bào)道的Ru基催化劑性能對(duì)比圖,凸顯了其領(lǐng)先地位。
(f-i) 長(zhǎng)期穩(wěn)定性測(cè)試表明,無論是在1 M KOH還是堿性海水中,催化劑經(jīng)過3000次CV循環(huán)和100小時(shí)的恒流測(cè)試后,性能幾乎沒有衰減。
圖4 原位光譜與同位素標(biāo)記揭示反應(yīng)機(jī)理: 直接證據(jù)表明活化的羥基參與了質(zhì)子交換過程。
-
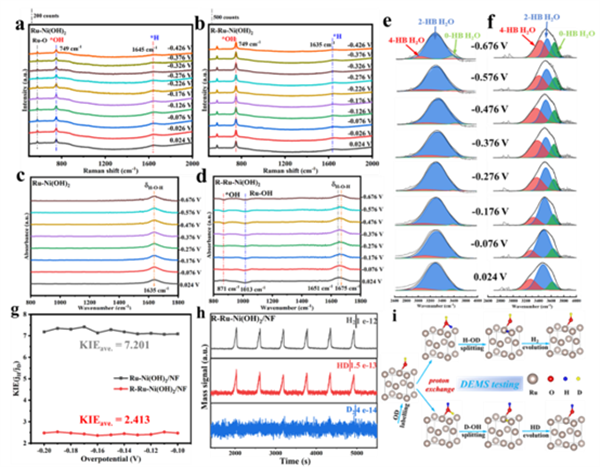
(a, b) 原位拉曼光譜顯示,活化后的R-Ru-Ni(OH)2在析氫電位下出現(xiàn)了更強(qiáng)的歸屬于Ru-OH的振動(dòng)峰,且隨電位負(fù)移而增強(qiáng),證明了OH物種參與了HER過程。
-
(c-f) 原位衰減全反射紅外光譜(ATR-SEIRAS)不僅捕捉到了OH中間體和Ru-OHad的信號(hào),還觀察到界面水的振動(dòng)峰發(fā)生紅移,表明活化后的表面促進(jìn)了界面水分子的解離。
(g) 動(dòng)力學(xué)同位素效應(yīng)(KIE)測(cè)試表明,活化后催化劑的KIE值顯著降低(從7.201降至2.413),意味著O-H鍵的斷裂(即水解離步驟)更容易發(fā)生。
(h, i) 關(guān)鍵的同位素標(biāo)記-微分電化學(xué)質(zhì)譜(DEMS)實(shí)驗(yàn):用D?O活化催化劑引入ODad后,在H2O電解液中進(jìn)行HER,檢測(cè)到了大量的HD氣體信號(hào)。這直接證明了表面的D物種(源于ODad)參與了反應(yīng),是通過與H2O中的H進(jìn)行交換后生成的。
圖5 密度泛函理論(DFT)計(jì)算: 從理論上闡明了羥基促進(jìn)HER的內(nèi)在機(jī)制。
-

(a) 提出了活化前后反應(yīng)路徑的示意圖,清晰展示了“質(zhì)子交換”這一核心機(jī)制。
-
(b-g) 電子結(jié)構(gòu)計(jì)算(功函數(shù)、態(tài)密度、差分電荷密度、電子定域函數(shù))表明,活化引入的OHad優(yōu)化了Ru位點(diǎn)的電子結(jié)構(gòu)(功函數(shù)降低、d帶中心下移、電荷重新分布),使其更容易進(jìn)行電子轉(zhuǎn)移和質(zhì)子捕獲。
(h, i) 吉布斯自由能和能壘計(jì)算是理論部分的核心。結(jié)果表明:1)有OHad參與的質(zhì)子交換路徑(能壘0.193 eV)在能量上遠(yuǎn)優(yōu)于傳統(tǒng)的水吸附路徑;2)OHad的存在顯著降低了后續(xù)水解離(能壘僅0.035 eV)和H脫附(能壘0.395 eV)的能壘。
小結(jié): 理論計(jì)算完美支撐了實(shí)驗(yàn)結(jié)論,證實(shí)了OHad通過開辟一條低能壘的“質(zhì)子交換”新路徑,雙向加速了水的吸附/解離和H2的脫附,從而根本上提升了催化活性。
最后點(diǎn)評(píng)
這絕對(duì)是一項(xiàng)構(gòu)思巧妙且論證嚴(yán)謹(jǐn)?shù)慕艹龉ぷ鳌Kㄟ^一個(gè)簡(jiǎn)單的電化學(xué)活化策略,顛覆了堿性HER研究中對(duì)表面羥基(OH)作用的傳統(tǒng)認(rèn)知——即OH不再僅僅是阻礙反應(yīng)的“毒物”,而是可以被巧妙利用的“助推器”。
文章最大的亮點(diǎn)在于其提出的“羥基-界面水質(zhì)子交換”機(jī)理。作者結(jié)合了多種強(qiáng)大的原位光譜技術(shù)和關(guān)鍵的同位素標(biāo)記DEMS實(shí)驗(yàn),層層遞進(jìn),提供了無可辯駁的證據(jù),令人信服地揭示了活化羥基(OHad)如何作為質(zhì)子受體,主動(dòng)參與并促進(jìn)水分子吸附、解離和最終的氫氣脫附。DFT計(jì)算不僅在理論層面驗(yàn)證了這一新路徑的能量?jī)?yōu)勢(shì),還從電子結(jié)構(gòu)層面深刻揭示了其內(nèi)在原因。
這項(xiàng)工作不僅為開發(fā)高性能堿性HER催化劑提供了一種高效且普適的改性策略,更重要的是,它為我們從原子和分子層面深入理解復(fù)雜電催化反應(yīng)機(jī)理開辟了新的視角。它告訴我們,優(yōu)化催化劑的設(shè)計(jì)思路可以更加靈活,有時(shí),轉(zhuǎn)化甚至利用那些被認(rèn)為是“不利”的中間物種,可能會(huì)帶來意想不到的突破。
文獻(xiàn)引用:
[1] Jiang, L.; Chen, X.; Jiang, L.; Luo, X.; Li, R.; Zhou, Q.; Mu, X.; Chen, L.; Li, B.; Yu, J.; Mu, S. Hydroxyl?Interfacial Water Proton Exchange Strategy to Hasten Water Reduction on Ruthenium Sites. ACS Catal. 2025, 15, 14380–14391.
文章鏈接:https://doi.org/10.1021/acscatal.4c07602